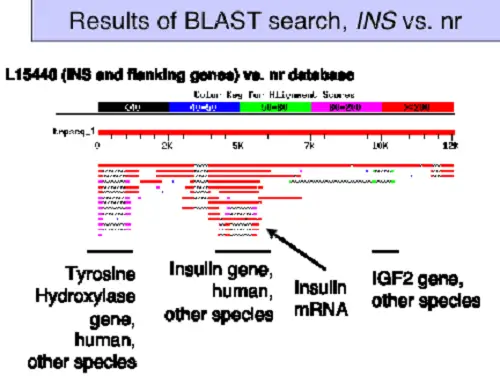
BLAST and FASTA are bioinformatic tools[i] used to compare protein and DNA sequences for similarities that mostly arise from common genetics. Both programs use a score strategy to do comparisons between the sequences, producing highly accurate results.
In molecular biology, two sequences are compared to evaluate structure and function to determine if the sequences are related.
A sequence is a collection of connected nucleotides or amino acid residues. A DNA sequence consists of a collection of connected nucleotides, and a protein sequence consists of the amino acids.
Sequencing determines the nucleotide sequence in a DNA sequence, or an amino acid sequence in a protein. Several methods are available with institutes like the National Center of Biotechnology Information Institute (NCBI), where deposits of sequences are available for searching.
Local and Global Sequence Alignments
A sequence alignment is a sequence comparison, which is the basis of bioinformatics. The sequence describes the DNA or protein in order to find the similarities, and any structural and functional relationship between two sequences.
An alignment finds the similarity by comparing the query sequence with the sequences in the databases. The algorithm finds the alignment by assigning scores, and the greater the similarities, the greater the chance for both sequences to have a structural or functional relationship.
Two main methods of sequence alignment:
- Local alignment finds the local regions with a high level of similarity and the alignment is performed between two nucleotide or amino acid sequences to identify structural or functional similarities.
- Global alignment is performed from beginning to end of the sequence to find the best possible alignment.
The local and global alignment use different algorithms and a score matrix.
The scoring matrix assigns a positive/or higher value for a match, and a negative or lower value for a mismatch.
Similarity searches are run between a query sequence and the sequences database (e.g NCBI), and the detected genes in the sequence are matched to similar sequences retained in the database.
BLAST
BLAST is an acronym that stands for Basic Local Alignment Search Tool, which uses local sequence alignment searching.
It is an algorithm used to compare biological sequences, predominantly for protein searches, and a well-known, and widely used tools in bioinformatics. It is noted for its ability to accurately (and very quickly) identify similarities between two local sequence alignments by calculating an expected value and then estimating the amount of matches for the two sequences.
Proteins (most) are modular with functional and structural sequences within the protein. The same sequences could occur in proteins from different species, and BLAST is designed to find these similar sequences.
For example, comparing the insulin protein (amino acid) sequences between a rat and a guinea pig, both proteins sequences are aligned to find the percent of similarity between the two.
BLAST has also been tweaked over time producing several variant searches, namely, megaBLAST, BLASTN, BLASTP (the basis of other BLAST types like BLASTX and TBLASTN), PSI-BLAST, RPSBLAST, and DELTA-BLAST searches.
The BLAST algorithm reads the query search parameters, searches the database, and produces a set of patterns used to initiate matched sequences. Several actions occur for each sequence in the database when scanning the matches to patterns, which initiates a gap-free extension that has a specific score.
A “gap” indicates an amino acid residue could have been deleted from the sequence or one or more residues inserted into the second sequence. Gapped extensions with a minmum score achieved are saved. Lower scoring matches could be remove if too many matches come up. Finally, in the last phase (known as a traceback), the insertions and deletions are calculated using sensitive parameters.
FASTA
FASTA is a DNA sequence alignment software package that also uses local sequence first, but then extends to global alignment, and is best used for nucleotide searches (DNA comparisons).
FASTA is used to search similarities between sequences of DNA and highly regarded for similarity searching. The FASTA format is ever present in bioinformatics and a widely used input format for other tools such as BLAST.
The query sequence is organized into sequence patterns (k-tuples). The target sequence is searched to find these k-tuples and any similarities. It is of popular opinion to conduct the full search by first running a BLAST search, then FASTA.
The FASTA file format is widely used as the input method in other sequence alignment tools like BLAST.
The estimates from BLAST and FAST are very reliable, however in rare cases, estimations do fail. When an unexpected alignment occurs, scientists should note the significance and take on further steps to analyze and run additional searches with variations of the original sequences.
Author: Gillian Douglas
Content Developer specialized in video production and editing, creative and technical writing, logo and web design, UI design and software testing.









Leave a Reply